Kobber er et metall som har mange viktige funksjoner i kroppen vår. Metabolismen av andre næringsstoffer, deriblant jern, er avhengig av en godt regulert kobbermetabolisme for å fungere optimalt. Kobber er etter hvert satt i sammenheng med funksjon og utvikling av nervesystemet, hjertefunksjon, beinmetabolismen og immunsystemet. Mennesker inneholder omtrent 1mg kobber per kg kroppsvekt, og ettersom vi ikke har noen lagre av metallet så er vi avhengig av å få det i oss jevnlig gjennom kostholdet. Vi finner kobber i to oksidasjonsstadier, Cu+ og Cu2+.

Bilde: Colourbox
Denne artikkelen hører til artikkelserien om næringsstoffene.
Opptak og metabolisme
Kobber absorberes hovedsakelig i tynntarmen, men noe absorberes også i magesekken. En kobbertransporter som heter Ctr1 står for den aktive absorbsjonen, mens passiv diffusjon er antatt å spille en rolle dersom inntaket blir stort. Inne i tarmcellene kan kobber enten brukes lokalt, eller fraktes videre i blodet til leveren (noe tas også opp av nyrene). Dette styres av såkalte kobberchaperoner, som er proteiner som frakter kobberet rundt inne i cellen. Disse er beskrevet mer i detalj lenger nede. I blodet fraktes kobber hovedsakelig bundet til albumin og α2-globulin til leveren, mens fra leveren fraktes kobber først og fremst bundet til ceruloplasmin. De som mangler ceruloplasmin viser imidlertid ikke noen tegn til forstyrrelser i kobbermetabolismen, noe som tyder på at kobber også fraktes fra leveren bundet til andre molekyler. Dette er ikke fullstendig kartlagt.
Opptaket av kobber er regulert for å tilpasses kroppens behov. For å kunne absorberes gjennom Ctr1 må kobberet først reduseres til Cu+. I tillegg har cellene et komplekst nettverk av kobbertransportproteiner som sørger for å transportere kobberet dit det skal i de mengdene som trengs. Overflødig kobber kan binde til metallothionein, noe som antas å være en forsvarsmekanisme mot kobberforgiftning, en slags intracellulær detoksifiseringsmekanisme.
Fra blodet tas kobber opp i cellene gjennom det samme transportproteinet som vi fant i tarmen, Ctr1. Inne i cellene binder kobberet til såkalte chaperoner. Dette sørger for at konsentrasjonene av fritt kobber holdes lav, noe som reduserer risikoen for toksiske effekter. Vi har hovedsakelig tre slike chaperoner, som hver leverer kobber til spesifikke steder i cellen. CCS er et chaperon som frakter kobber til enzymet superoksid dismutase, som er en viktig antioksidant. Cox17 leverer kobber til mitokondriene for inkorporering i cytokrom C oksidase, som er et sentralt enzym i elektrontransportkjeden. Du kan lese mer om disse i avsnittet om funksjoner. Det siste chaperonet heter Atox1, og leverer kobber til noen energikrevende kobbertransportproteiner, ATPaser, som transporterer kobber inn i golginettverket, der kobberet kan kobles til proteiner og sendes ut i blodet eller skilles ut fra kroppen. Disse transportproteinene heter ATP7A og ATP7B.
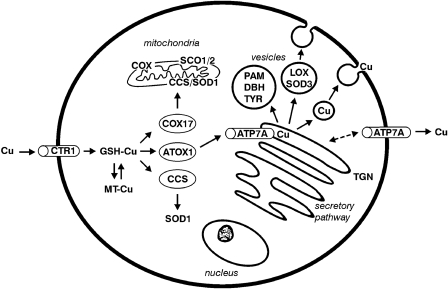
Tumer Z, Moller LB: Menkes disease. Eur J Hum Genet 2010, 18(5):511-518.
ATP7A er uttrykt i alle vev utenom leveren. Dette proteinet transporterer kobber fra cellenes cytosol og inn i golginettverket. Her kan kobberet inngå i vevsspesifikke enzymer, og dermed utføre sin funksjon. I tarmcellene er det ATP7A som frakter kobber over i blodet slik at det kan fraktes til leveren. I leveren har vi ikke ATP7A, men et tilsvarende protein som heter ATP7B. Dette proteinet sørger for at kobber kan kobles til ceruloplasmin, eller eventuelt skilles ut i gallen. Noen nerveceller inneholder også ATP7B. Disse ATPasene regulerer kobbermetabolismen.
Det er to genetiske sykdommer som rammer kobbermetabolismen. Menkes sykdom skyldes en defekt i ATP7A, noe som fører til en systemisk kobbermangel. Dette skyldes at kobber ikke fraktes fra tarmcellene og over i blodet, samtidig som ATP7A også er ansvarlig for å overføre kobber til en rekke av de kobberavhengige enzymene. Wilsons sykdom skyldes en defekt i ATP7B, og fører til kobberoverbelastning i leveren. Dette er fordi det er ATP7B som sørger for å frakte kobber fra leveren og ut i blodet eller skille det ut i gallen, og mangel på dette proteinet gjør at kobber hoper seg opp. Mer informasjon om Menkes sykdom finner du i denne artikkelen.
Funksjoner
Kobber er viktig for funksjonen til en rekke enzymer, og er innblandet i syntese av bindevev, jernmetabolismen, energiproduksjon, immunfunksjon og i nervesystemet. Kobberavhengige enzymer fungerer ved å binde oksygen. I denne prosessen omdannes kobberet mellom de to formene, Cu(I) og Cu(II), og det produseres forskjellige organiske produkter. Som nevnt under avsnittene om metabolisme så har vi tre kobberchaperoner som frakter kobberet til de ulike enzymene.
- Amin oksidaser
En gruppe kobberavhengige enzymer er amin oksidasene. Disse deaminerer monoaminer (monoamin oksidase) og diaminer (diamin oksidase). Disse enzymene har en funksjon i metabolismen til enkelte aminer, og er blant annet ansvarlig for deaktivering og nedbrytning av fysiologisk aktive aminer som histamin. I tillegg har de en funksjon i intracellulær signaloverføring ved å produsere hydrogenperoksid.
- Ceruloplasmin
Mesteparten av kobberet i blodet er bundet til ceruloplasmin, som er et kobberavhengig protein som produseres i leveren. Ceruloplasmin er også kjent som ferroksidase, og har en viktig funksjon i jernmetabolismen ved å redusere jern slik at det kan binde til transferrin. Mangel på ceruloplasmin påvirker ikke kobbermetabolismen i seg selv, men hindrer at jern binder til transferrin. Dette fører til at jern må binde til andre molekyler, noe som kan gi jernoverbelastning. Hephaestin er et membranbundet kobberavhengig protein som også kan redusere jern. Nylig er det også oppdaget enda en ferroksidase, zyklopen, som man tror har som funksjon å regulere transport av jern over morkaken til fosteret.
- Cytokrom C oksidase
Cytokrom C oksidase (CCO) er et viktig proteinkompleks som er involvert i energimetabolismen ved å inngå i elektrontransportkjeden (kompleks IV). Dette komplekset er med å bygge opp protongradienten som er viktig for å kunne produsere energimolekylet ATP. Komplekset er også avhengig av jern, sink og magnesium for å fungere. Aktiviteten til CCO er høyest i hjertet, og er også høy i hjernen, leveren og nyrene.
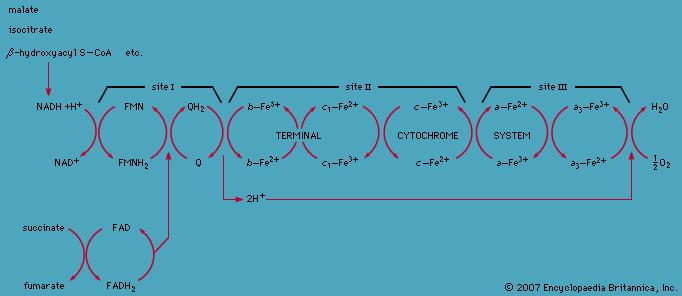
Skjematisk oversikt over elektrontransportkjeden. Bilde: www.britannica.com
- Dopamin-β-monooksygenase
Dopamin-β-monooksygenase (DBM) er enzymet som produserer noradrenalin fra dopamin. Dette enzymet finner vi i binyremargen, sympatiske nerveceller i det perifere nervesystemet og i hjernens adrenerge og noradrenerge nevroner. DBM er avhengig av C-vitamin som kofaktor.
- Superoksid dismutase
Superoksid dismutase (SOD) er en viktig antioksidant som tar seg av det reaktive oksygenmolekylet superoksid. Vi har tre varianter av SOD, og to av disse er kobberavhengige, SOD1 og ekstracellulær SOD. Begge disse er også avhengig av sink som en kofaktor.
- Lysyl hydroksylase
Kobber er også viktig for syntese og stabilisering av bindevev. Enzymet lysyl hydroksylase (LO) omdanner aminosyren lysin i nydannet bindevev til hydroksylysin, som er viktig for at flere kollagentråder skal kunne binde seg til hverandre og dermed bli sterkere. Dette er også viktig for styrken til beinvev, blodårevegger, lunger, hud og tenner.
- Tyrosinase
Tyrosinase er et kobberavhengig enzym som er viktig for pigmentering av huden gjennom å katalysere produksjonen av melanin. De eksplisitte funksjonene til tyrosinase i denne sammenhengen er å gjøre tyrosin om til dopamin, og deretter oksidere dopamin til dopakinon. Mangel på tyrosinase gir albinisme.
- Peptidylglycin α-amiderende monooksygenase
Peptidylglycin α-amiderende monooksygenase (PAM) er et viktig enzym som sørger for posttranslasjonell modifisering av en rekke peptider slik at de blir bioaktive. PAM er blant annet viktig for aktivering av over halvparten av nevropeptidene våre, deriblant kalsitonin (beinhelse) og kolecystokinin/gastrin/nevropeptid Y (sult- og metthetsregulering). PAM er avhengig av C-vitamin som kofaktor, og vi finner mye av dette enzymet i nervevev og endokrine celler, og spesielt høye konsentrasjoner finner vi i hypofysen og i hjertet.
Behov og anbefalinger
Det er beregnet at et inntak i området 0,7mg/dag vil være tilstrekkelig for voksne. Et gjennomsnittlig kosthold gir 1-2mg/dag.
Mangel
Kobbermangel er veldig sjelden ettersom homeostasen er så godt regulert som den er. Det er likevel mulighet for å utvikle mangel ved veldig lavt inntak over lengre tid. Mangel vil enten oppstå som følge av for liten tilførsel eller for stor utskillelse. De som er i risikosonen for å utvikle mangel er de som i lengre tid er avhengig av parenteral ernæring (intravenøs ernæring), premature barn som ikke får tilført nok (i denne perioden bygges det opp et lite kobberlager i leveren som følge av manglende evne til utskillelse), små barn med kronisk diaré, personer med store brannskader eller de som får i seg for mye sink (hemmer opptaket av kobber). Sykdommer som gir malabsorbsjon, som cøliaki, cystisk fibrose, inflammatorisk tarm eller kirurgisk fjerning av tarm, kan også føre til kobbermangel. Den genetiske sykdommen Menkes sykdom gir kobbermangel som følge av defekt ATP7A.
Vanlige kjennetegn ved kobbermangel er hypokrom anemi, hypopigmentering, neutropeni, trombocytopeni og dysfunksjon i skjelettet, sirkulasjonssystemet og immunsystemet. Kobber er også viktig for utvikling og funksjon av nervesystemet, så mangel kan gå hardt utover dette.
Hypokrom anemi kjennetegnes ved at det dannes for få røde blodceller, og disse inneholder også mindre hemoglobin enn vanlig. Blodets kapasitet for å frakte oksygen vil derfor være nedsatt. Denne anemien vil ikke respondere på økt inntak av jern. Mekanismene som ligger til grunn er ikke fullstendig kartlagt, men det ser ut som om hovedproblemet er at man ikke klarer å utnytte jernet slik man skal. Forstyrrelser i utviklingen av blodceller er også antatt å være grunnen til at det dannes for lite hvite blodceller (neutropeni) og blodplater (trombocytopeni).
I dyrestudier har man sett dysfunksjon i hjertet som følge av lavt kobberinntak, og det er antatt at dette skyldes redusert aktivitet av kobberavhengige enzymer. Dette har gitt grunnlag for spekulasjoner om kobbermangel kan spille en rolle i utviklingen av hjerteinfarkt hos mennesker, og studiene har gitt sprikende resultater. Mangelfull aktivitet i LO-enzymet kan også ramme bindevevet som omkranser blodårene, og mangelfull antioksidantfunksjon kan også spille en rolle for den kardiovaskulære helsen. Ved kobbermangel ser man ofte også forstyrrelser i blodlipidene med høye triglyserider og LDL-kolesterol.
Sammenhengen mellom kobbermangel og redusert beinhelse skyldes redusert aktivitet i LO. Dette gir svakheter i bindevevet, og er assosiert med beinskjørhet. Disse symptomene minner om det man ser ved skjørbuk (c-vitaminmangel), noe som er logisk ettersom LO også er avhengig av C-vitamin. Det er indikasjoner på at kobbertilskudd kan redusere beintap, men hvorvidt tilskudd hos eldre vil ha en klinisk viktig effekt på beinhelsen gjenstår å se.
Kobbermangel er satt i sammenheng med økt risiko for å rammes av infeksjoner. Dette skyldes både redusert produksjon av immunceller og redusert funksjon av de immuncellene som produseres. Kobbermangel ser ut til å redusere produksjonen av proinflammatoriske cytokiner som er viktige for å bekjempe en infeksjon. Linken mellom nedsatt funksjon i immuncellene og kobbermangel er ikke veldig sterk, men mye tyder på at det er en sammenheng.
Sent i fosterutviklingen og under ammingen bygger det seg opp et kobberlager hos fosteret/barnet. Mangelfullt inntak hos mor i disse periodene kan derfor slå negativt ut for barnet, og da spesielt utviklingen av nervesystemet. Kobbermangel gir redusert funksjon av en rekke kobberavhengige enzymer, og mange av disse er viktig for funksjonen til nervesystemet. Fokuset så langt har vært på kobber sin rolle i utviklingen av nervesystemet, men det er også identifisert funksjoner som er avhengig av tilstrekkelig tilførsel hele livet. Flere tilfeller av myelopati er satt i sammenheng med kobbermangel.
Kan vi få for mye?
Som med mangel så gjør de homeostatiske mekanismene at kobberforgiftning er uhyre sjelden. Det kan derimot under visse omstendigheter skje, for eksempel dersom man får i seg vann som er forurenset med kobber eller spiser syrlig mat som har vært oppbevart i kobberholdige bokser. Små barn er ekstra utsatt ettersom deres evne til å skille ut kobber ikke er fullt utviklet.
Kobberforgiftning kan gi økt oksidativt stress, ettersom kobber i likhet med jern kan inngå i Fentonreaksjoner som produserer reaktive oksygenforbindelser. Symptomer på akutt kobberforgiftning er oppkast og gastrointestinale plager.
Den genetiske sykdommen Wilsons sykdom gir kobberopphopning i leveren og dermed forgiftning.
Biotilgjengelighet og gode kilder

Bilde: Colourbox
Hvor godt vi absorberer kobber vil variere med hvor mye vi får i oss. Enkelte proteiner, sink, jern og c-vitamin kan ha en negativ effekt på biotilgjengeligheten.
De viktigste kildene til kobber i kostholdet vårt er vegetabilske, og da spesielt nøtter, frø og fullkorn. Skalldyr, organkjøtt og sjokolade bidrar også. Drikkevann kan i noen tilfeller være en kilde, spesielt dersom det fraktes gjennom kobberrør.
Aktuelt om kobber
- Grunnet at mange kobberproteiner er uttrykt i hjernen er kobber satt i sammenheng med en rekke nevrologiske sykdommer.
Artikkelen er sist oppdatert juli 2013
Tilbake til artikkelserien om næringsstoffene.
[…] http://www.friskogfunksjonell.no/kobber/ […]